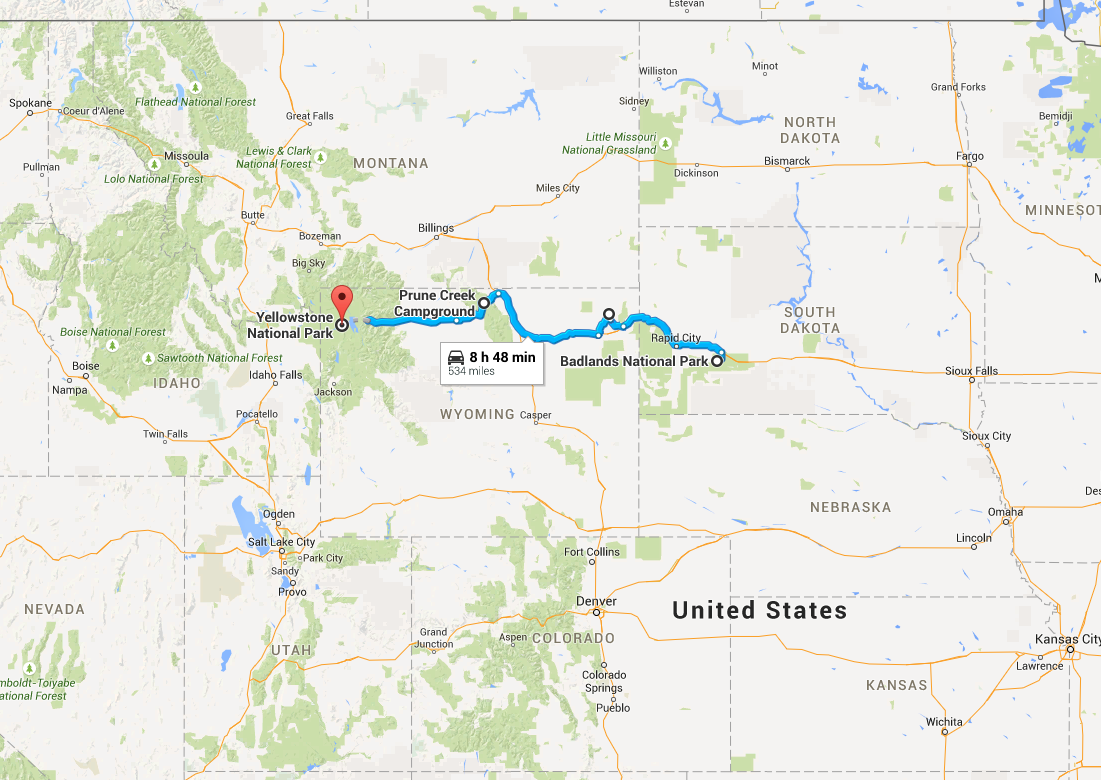
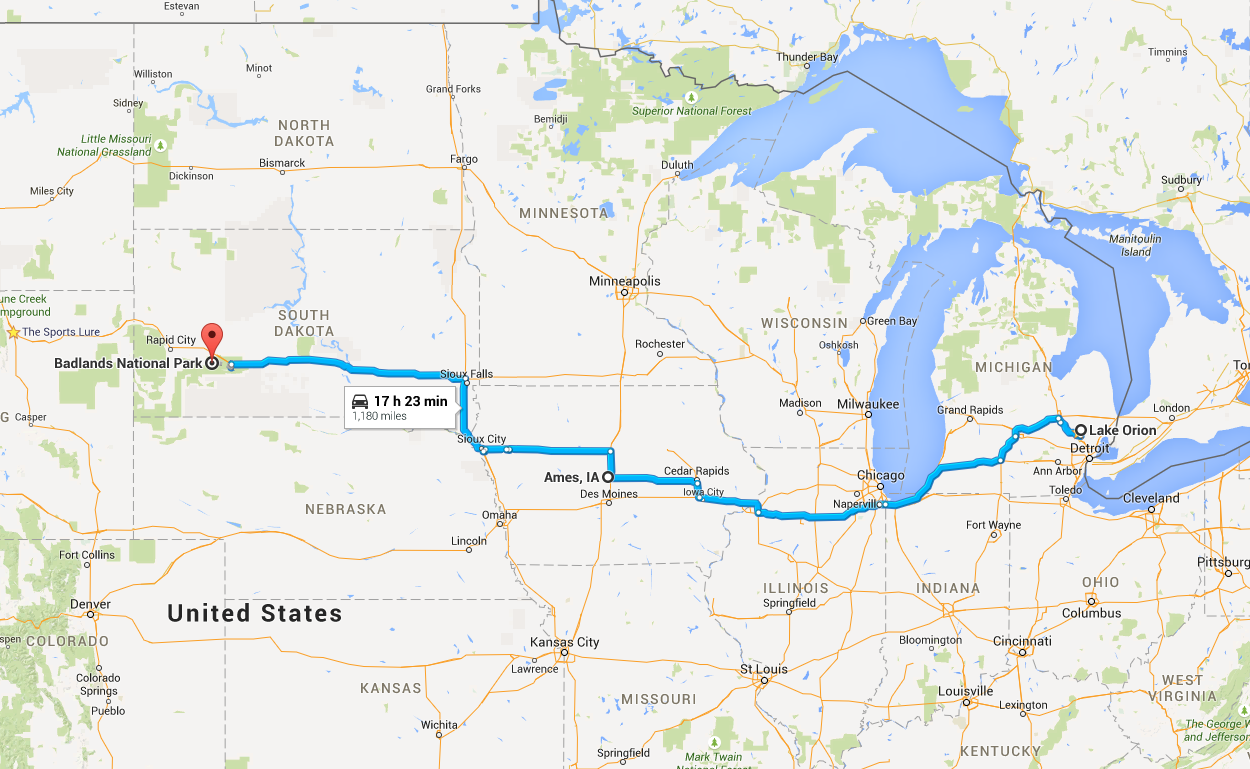
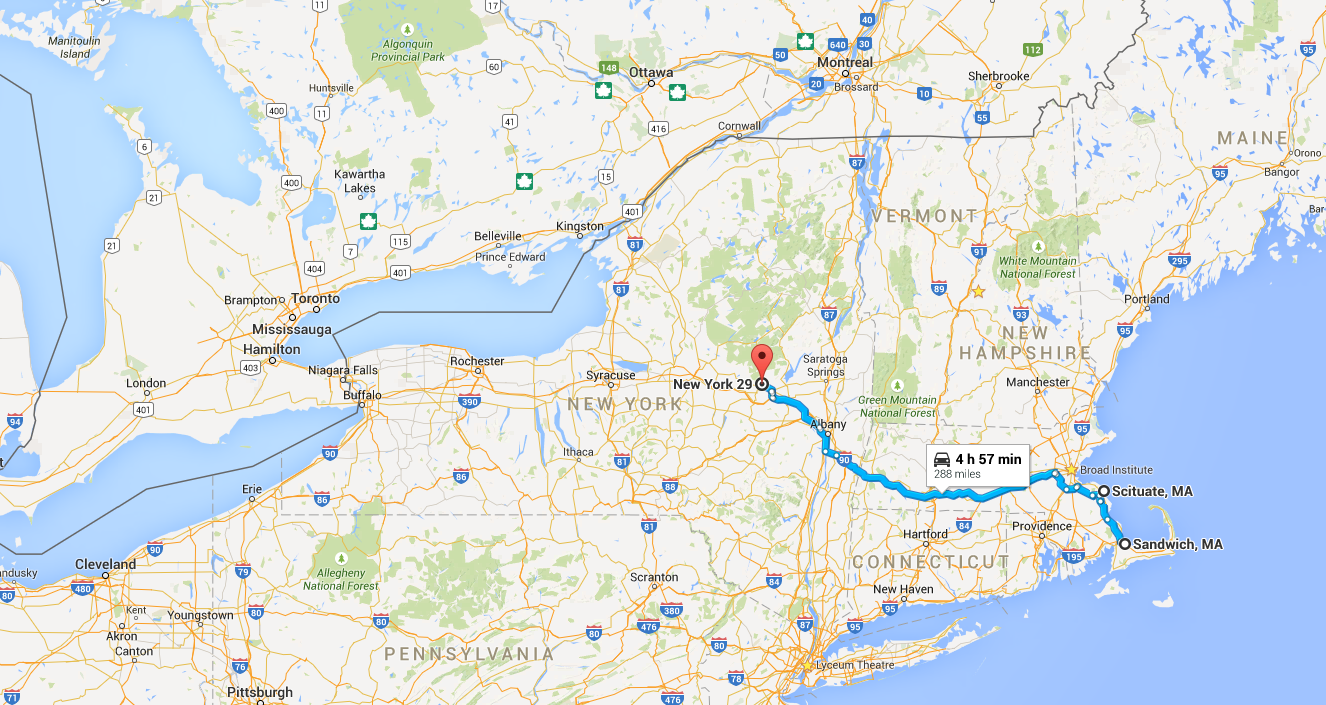
My girlfriend Lizzy flew out to Seattle to join me for the next section of the trip – a tour down the West Coast. Our first day in the city was packed with visiting the interesting sites in downtown and West Seattle (where we were staying in an airbnb).
First stop: Pike Place Market. The famous indoor/outdoor market spans several blocks by the water in downtown Seattle. You’re immediately greeted by stalls of fresh fish, produce, craft-made everything. I think I became fifty bucks poorer just walking onto Pike street. Just look at this fish stall!

We explored the market and stocked up on provisions for the next sections of the trip. Buckwheat honey (surprisingly good on oats), salmon jerky and Italian salami should drive away the mundane tastes of backpacking food. I also visited a few coffee shops to try some of Seattle’s famous brews, and eventually decided to take home a bag from Seattle Coffee Works. It tastes great in the new traveling french press I also picked up!
Downtown had some more interesting sights to check out, like the Chihuly Garden and Glass museum. Most of Dale Chihuly’s work is displayed in this indoor-outdoor museum. The color and scale of some of the exhibits are truly stunning.


One thing I immediately noticed about Seattle in contrast to the East Coast: people are friendly out here. Not just fake “how’s it going” friendly. No. Actually friendly. Interested in talking to strangers and learning their story. I had a 20 minute conversation about traveling with a guy on a beach in West Seattle. Lizzy and I talked about farming and produce to a vendor in Pike Place, after which he gave us a great deal on the food we had lined up. I just can’t see these conversations happening in Boston or Providence. Time to start looking at grad schools in Washington? Maybe…