In the microbiome field we struggle with the fact that reference databases are (sometimes woefully) incomplete. Many gut microbes are difficult to isolate and culture in the lab or simply haven’t been sampled frequently enough for us to study. The problem is especially bad when studying microbiome samples from non-Western individuals.
To subvert the difficulty in culturing new organisms, researchers try to create new reference genomes directly from metagenomic samples. This typically uses metagenomic assembly and binning. Although you most likely end up with a sequence that isn’t entirely representative of the organism, these Metagenome Assembled Genomes (MAGs) are a good place to start. They provide new reference genomes for classification and association testing, and start to explain what’s in the microbial “dark matter” from a metagenomic sample.
2019 has been a good year for MAGs. Three high profile papers highlighting MAG collections were published in the last few months[1,2,3]. The main idea in each of them was similar – gather a ton of microbiome data, assemble and bin contigs, filter for quality and undiscovered genomes, do some analysis of the results. My main complaint about all three papers is that they use reduced quality metrics, not following the standards set in Bowers et al. (2017). They rarely find 16S rRNA sequences in genomes called “high quality,” for example.
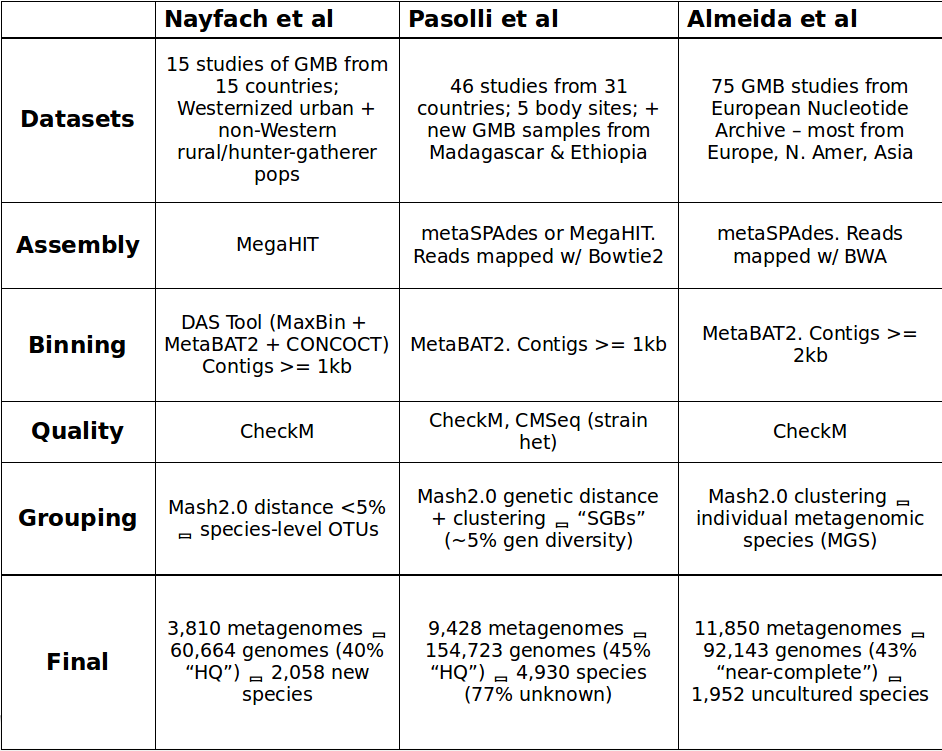
Comparing the datasets, methods, and results from the three MAG studies. This table was compiled by Yiran Liu during her Bhatt lab rotation.
After reading the three MAG papers, Nayfach et al. (2019) is my favortie. His paper does the most analysis into what these new genomes _mean_, including a great finding presented in Figure 4. These new references assembled from metagenomes can help explain why previous studies looking for associations between the microbiome and disease have come up negative. This can also help explain why microbiome studies have been difficult to replicate. If a significant association is hiding in these previously unclassified genomes, a false positive association could easily look significant because everything is tested with relative abundance.
In the Bhatt lab, we were interested in using these new MAG databases to improve classification rates in some samples from South African individuals. First we had to build a Kraken2 database for the MAG collections. If you’re interested in how to do this, I have an instructional example over at the Kraken2 classification GitHub. For samples from Western individuals, the classification percentages don’t increase much with MAG databases, in line with what we would expect. For samples from South African individuals, the gain is sizeable. We see the greatest increase in classification percentages by using the Almeida et al. (2019) genomes. This collection is the largest, and may represent a sensitivity/specificity tradeoff. The percentages represented below for MAG databases are calculated as the total classifies percentages when the unclassified reads from our standard Kraken2 database are run through the MAG database.

Classification percentages on samples from Western individuals. We’re already doing pretty good without the MAG database.
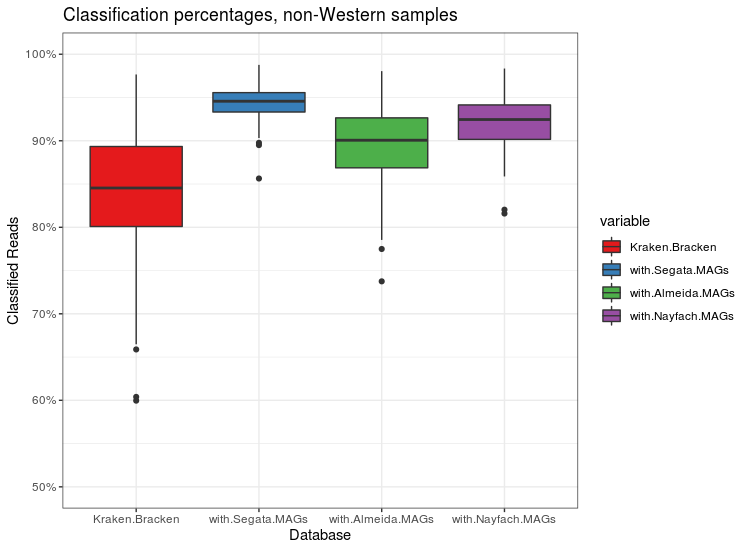
Classification percentages on non-Western individuals. MAGs add a good amount here. Data collected and processed by Fiona Tamburini.
References
1.Nayfach, S., Shi, Z. J., Seshadri, R., Pollard, K. S. & Kyrpides, N. C. New insights from uncultivated genomes of the global human gut microbiome. Nature 568, 505 (2019).
2.Pasolli, E. et al. Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle. Cell 0, (2019).
3.Almeida, A. et al. A new genomic blueprint of the human gut microbiota. Nature 1 (2019). doi:10.1038/s41586-019-0965-1
4.Bowers, R. M. et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nature Biotechnology 35, 725–731 (2017).